�l���r�g��2020-04-29���ٷ���t�WՓ���g�[��1��
ժ Ҫ�� ժҪ�քe��1,6-�������͌���������ԭ�ϣ��c��ӳ�w��L-�i����l���s���M���õ��������ӻ�������ϳɰ����Ӽ����������g�a�{�^�߷ֱ��|�V���˴Ź�����V�ͺ˴Ź���̼�V���M�б������_�J�Y�������⣬������2A��7B-1ͨ�^X-ray�ξ����䷨�_���˽Y���� �P�I
����ժҪ�քe��1,6-�������͌���������ԭ�ϣ��c��ӳ�w��L-�i����l���s���M���õ��������ӻ�������ϳɰ����Ӽ����������g�a�{�^�߷ֱ��|�V���˴Ź�����V�ͺ˴Ź���̼�V���M�б������_�J�Y�������⣬������2A��7B-1ͨ�^X-ray�ξ����䷨�_���˽Y����
�����P�I�~�����ӣ��ϳɣ��ξ�
����1.�����S��ˎ�ﻯ�W�IJ���lչ��ʹ�ô������F���Ժϳ��ѽ��ɞ�ʮ���ձ�ķ����������ֹ���^�ɽ��ٴ�����ø���ЙCС���Ӵ����ȱ��V��ʹ��[1][2][3]���e�ǽ������Ѹ�ٰlչ�����������ЙCС���Ӵ�����������������Ƃ䡢�r����������h���Ѻá������l���غ͵ȃ��ݶ��ܵ��ƌW�҂�����A���ɞ��о��ğ��c��ǰ��[4]��
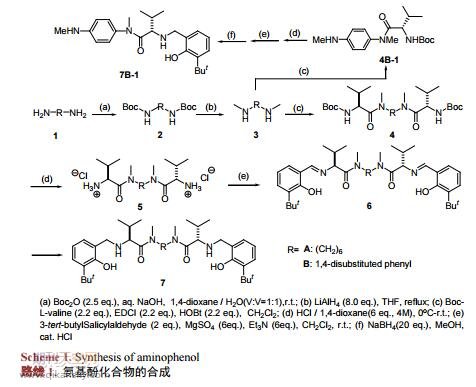
���������ӻ������Y���к��а��������u���ɂ����܈F�������H�����������w�c����Ti��Cu�Ƚj�Ϻ��c������Ҳ���ԆΪ�����һ�N�p�����ЙCС���Ӵ���ʹ�ã����ڴ�������h��������[5][6]�Լ�����[7][8]�IJ����Q�ӳɡ�Mannich����[9]��������ϩ����������[10][11][12]�ȶ�N�����ж����F�����ô����ԣ������������@���^�ߵĮa�ʺ͌�ӳ�x���ԡ�ͬ�r���@�������ͻ���ă��ݣ����ںϳɣ������ἃ�����������ٵȡ�������ϳ��·f�Y���İ����ӻ����������Ҫ�����x�������OӋ�ϳ������N�������ӻ�����(7A,7B,7B-1)��������7A��7B�քe�麬���ʽ�ͷ������Q�Y���İ����ӻ����������7B-1�麬�б����Y���İ����ӻ���������ӻ�����ĺϳ���·��1��ʾ��
����2.����
����2.1.��Ҫԇ�����x��
����INOVA-600�ͺ˴Ź���x(��TMS��Șˣ�CDCl3���܄�);BruckerEquinox55FT-TR�ͼt����V�x(KBr��Ƭ);Brucker���|�V�x(ESI��x���g);�͜،��ʹ��EYELAPSL-1810�ʹ�������͜غ��ˮ��(�oˮ�Ҵ�����|)��
�����������ԇ�������������ʯ���ѷг̞�60��~90�棬���У��oˮ�Ě�������cɰ�ж��Ԛ��w���o�¼ӟ�������ö�����ͪ�@ɫ;���������zG(���u��������˾);���ӌ������zGF254(���u��������˾);���ӌ�����(GF254����0.5%CMC����)���������(254nm)�^�y���õ������@ɫ��
����2.2.�����ӻ�����ĺϳ�
�����Ի�����7A�ĺϳɞ���[10]��
����50mL�A��ƿ�м��뻯����1A(0.23g,2mmol)�����ӻ���܄�(20mL,V1,4-�������h:Vˮ=1:1)����ʹ���ܽ���Һ����ԡ�l���·�������(Boc)2O(1.09g,5mmol,2.5equiv.)��ͬ�r�����μ�10%��NaOH��Һ��ʹPH������8~9֮�g���������Ӯ������Һ�Ҝؔ����^ҹ��TLC�O�y����������������(20mL×3)��ȡ��ˮ����10%��KHSO4��Һ�ữ��PH=2~3��������������ȡˮ�࣬�ϲ��ЙC�࣬�oˮNa2SO4����^�V���VҺ��s�õ��֮a��֮a�{���������ؽY�����û�����2A��
����50mL�A��ƿ�м���oˮTHF����ԡ�¼���LiAlH4(0.61g,16mmol,8.0equiv.)����������2A(0.63g,2mmol)��Ar���o�»���4h��TLC�O�y�������ڱ�ԡ�l���£���������Na2SO4ˮ��Һ��練�����^�V����������ϴ����w���ϲ��ЙC�࣬��s�ô֮a��p�����s����(b.p.70��,����44Pa)�ßoɫҺ�w3A��
����EDCI(0.17g,0.92mmol,2.2equiv.)��CH2Cl2(20mL)��HOBT(0.12g,0.92mmol,2.2equiv.)��Boc-L-�i����(0.20g,0.92mmol,2.2equiv.)������50mL�A��ƿ�У������ܽ⡣��3A(0.06g��0.42mmol)�ܽ���CH2Cl2�У���μ��뵽���������Һ�У��Ҝؔ��裬TLC�O�y�����������ꮅ���ڷ����wϵ�м���10wt%�ę�����(20mL)�����裬�������İ�ɫ�������^�V���VҺ��10wt%�ę�����(20mL)���NaHCO3���ʳ�}ˮϴ�죬�ϲ��ЙC�࣬�oˮNa2SO4����^�V����s�ô֮a�����������(PE:EA=2:1)�ßoɫ�͠�Һ�w4A��ֱ���M����һ��������
����50mL�A��ƿ�м���4A(0.13g��0.24mmol)����ԡ�����¼����}���1,4-�������h��Һ(4.0M,2.88mmol��0.72mL��12equiv.)�����������Ҝؔ��跴����1.5h���������wϵ�г���30min���⣬�p����s���õ��غ�ɫճ��Һ�w5A����������ֱ���M����һ��������
������Ᵽ�o�£��ں�5A��50mL�A��ƿ�м���3-�嶡��ˮ��ȩ(0.11g,0.6mmol,2.5equiv.)���oˮMgSO4(0.17g,1.44mmol,6.0equiv)��CH2Cl2(20mL)��Et3N(0.15g,1.44mmol,2.0mL��6.0equiv.)���Ҝؔ��跴���^ҹ��TLC�O�y�����������ꮅ�������Sɫ��Һ������ö̹��z���^�V�Գ�ȥMgSO4�����Ұ��}���}�����z����ʯ����/��������(2:1)ϴÓ����ϴҺ�oɫ��ϴÓҺ�p����s���Sɫ�͠������������ϴ���Sɫ�͠����Գ�ȥ���������Ұ��}���}���VҺ�ϲ����p����s�����Sɫ�͠�Һ�w6A��
�����ں��л�����6A(0.18g,0.27mmol)�ĈA�ן�ƿ�м���״�(25mL)����ԡ�������Ⱥ����NaBH4(0.10g,2.7mmol)��һ���}�ᣬ���S���w�a����ͬ�r��Һ���Sɫ׃��oɫ���^�m��ԡ����30min����������ϡ�}����Һ(2.0M)ֱ�������wϵpH<1���Գ�ȥ�����wϵ�ж����߀ԭ���������wϵ��CH2Cl2(20mL×3)��ȡ���ϲ��ЙC�࣬�oˮNa2SO4����^�V���p����s�����ù��w��CH2Cl2/������(1:8)�ؽY���û�����7A��
�������]��x��ˎ��W�t����ô�l��Փ��
����3.�Y���cӑՓ
����3.1.�����ļ�������
���������ļ����������ü�ȩ�������������̼����������״��Ȟ����ԇ�����F��������ԇ�����c�ķ����^�y�����چμ��������A�Ρ����ֱ�Ӽ�����������߀ԭ���ڲ����Ćμ��������^�õđ��á���Փ�IJ���Boc���o������ͨ�^����Boc2O���������������ٶȵȿɺܺõ����Ɔ�߅�������pBoc�ȸ��������õ��^�îa�ʵĻ�����2A������LiAlH4�仯�������ɷ�����ڲ���������һ�������õ�������3A��������2A�Ćξ�(�D1)��ͨ�^���������ؽY���@�á�������3A�ļ�����ͨ�^�p�����s(b.p.70��,44Pa)���F��
����3.2.�����ӻ�����ĺϳ�
������1,6-���������l���������cBoc2O���õ���������LiAlH4��������߀ԭ���ٰ��c�i����������������������ԗl��Ó��Boc�����x�����c�嶡��ˮ��ȩ�Ŀs�ϼ�߀ԭ�ȷ����ɳɹ��@�ð����ӻ�����7A��������1,4-����������ԭ�ϕr������ͬ���ϱȵėl���£����õ��˻�����4B��4B-1�⣬߀���x�����õ�������4B-2�����������C��[13][14]�����P�īI[15]�ķ����l�F(·��2)��������4B-2��L-�i������(3-���װ�������)-3-�һ�̼�������}���}(EDCI)��������c1-�u������������(HOBT)�Ŀs�Ϯa�������4B-1��4B-2���γɿ��������ڻ�����4B-1�Y���з��㰷���H�����^����ʹ���^�y�M�����g�w������4B-2�γɻ�����4B�����о��l�F��ͨ�^�������wϵ������һ�����ĉA�ɴ��M������4B-1����4B���D�������ͨ�^�{��L-�i��������������Ӱ������ɫ@���^�߮a�ʵĻ�����4B��4B-1����������4B��4B-1�M�к��m�������ɷքe�õ�������7B��7B-1��������7B-1�Ćξ�(�D2)��ͨ�^��ʯ����/���������Ļ���܄��ؽY���@�á�
����3.3.������2A��7B-1�ľ��w����
������R-AXISSPIDER������x�ό�������2A��7B-1�Ćξ��M�Мyԇ����ʯī��ɫ����MoKα(λ=0.71073Å)ݗ�䣬��������2A��153(2)K�ض�����2.64˚≤θ≤24.99˚��������ω-2θ���跽ʽ�ռ����䔵�������ռ���5447�������c��1562�����������c(R(int)=0.0499)������1420�����^�y�c[I>2σ(I)]���ھ��w�Y����������K��ֵ��������ܶȵ���߷��0.289e∙A−3����ͷ��−0.287e∙A−3����������7B-1����153(2)K�ض�����2.65˚≤θ≤27.5˚��������ω-2θ���跽ʽ�ռ����䔵�������ռ���12,770�������c��5123�����������c(R(int)=0.0357)������4778�����^�y�c[I>2σ(I)]���ھ��w�Y����������K��ֵ��������ܶȵ���߷��0.14e∙A−3����ͷ��−0.16e∙A−3��������2A��7B-1��ȫ�����Ȕ�������Lp����У���������������У�������w�Y����ֱ�ӷ������ȫ���ǚ�ԭ�ӵ����˼������ԅ�������С������������SHELXL-97����(�a��CIF�ij������Q)��F2�M�о��ޫ@�÷ǚ�ԭ�����˼������ԅ�������ԭ���ɲ�ֵFourier�ϳɺ���ՓӋ��õ������������˺���ͬ�Ԝض����Ӆ��c�Y��Ӌ�㣬�������c�����������w�W�������ڱ�1��
����4.���g�w���a�����
����4.1.������2A
������ɫ���w������89.2%;IR(KBr)/cm-1:3371(NH),1686(C=O);1HNMR(600MHz,CDCl3,ppm),δ:4.55(brs,2H,NH),3.03(m,4H,N-CH2),1.44(m,4H,CH2),1.42(s,18H,CH3),1.31(m,4H,CH2);13CNMR(150MHz,CDCl3,ppm),δ:156.1(C=O),79.1(CtBu),40.5(N-CH2),30.1(CH2),28.5(CH3tBu),26.4(CH2).HRMS(ESI)Calcd.forC16H32N2O4(M+H+):317.2396,Found:317.2537.131
����4.2.������3A
�����oɫҺ�w������65.8%;1HNMR(600MHz,CDCl3,ppm),δ:2.45(m,4H,N-CH2),2.32(m,6H,N-CH3),1.38(m,4H,CH2),1.24(m,4H,CH2),0.89(brs,2H,NH);13CNMR(150MHz,CDCl3,ppm),δ:52.1(N-CH2),36.6(N-CH3),29.9(CH2),27.3(CH2).HRMS(ESI)Calcd.forC8H20N2(M+H+):145.1660,Found:145.1717.
����4.3.������6A
�������Sɫ�͠�Һ�w������58.6%;1HNMR(600MHz,CDCl3,ppm),δ:13.55(brs,2H,OH),7.99(s,2H,N-CH),7.31(d,2H,J=7.6Hz,Ar-H),7.20(s,4H,Ar-H),6.98(d,2H,J=6.0Hz,Ar-H),6.78(t,2H,J=7.6Hz,Ar-H),3.67(d,2H,J=7.6Hz,CH=N),3.31(S,6H,N-CH3),2.43(m,2H,CHiPr),1.41(s,18H,CH3),0.96(d,6H,J=4.4Hz,CH3iPr),0.82(d,6H,J=4.0Hz,CH3iPr);HRMS(ESI)Calcd.forC40H62N4O4(M+H+):663.4805,Found:663.4641.
SCISSCIAHCI